Forum
Warto wiedzieć
Twoje Forum
Forum Giełda
+Dodaj wątek
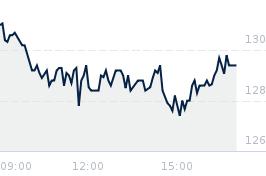
Opublikowano przy kursie:
30,30 zł
, zmiana od tamtej pory:
800,99%
Re: EVR- wycena 29 zł +67ze kardioznacznik
Zgłoś do moderatoraIII Faza
III faza badań klinicznych trwa od roku do kilku lat, i jest prowadzona z udziałem aż kilku tysięcy chorych (od 1 000 do 3 000 i więcej). Pacjenci kwalifikowani do badań III fazy to chorzy ze wskazaniem do farmakoterapii, kwalifikowani są do badania na podstawie ścisłych kryteriów mających na celu przebadanie leku w populacji zbliżonej do populacji chorych występującej w praktyce medycznej. Prowadzona jest ona zwykle przez wiele ośrodków klinicznych, często w wielu krajach. Liczba pacjentów włączanych do badania uwarunkowana jest wymogami rejestracyjnymi (FDA lub EMEA) oraz statystycznymi (szacowana wielkość próby).
Faza III ma na celu ostateczne potwierdzenie skuteczności badanej substancji w leczeniu danej choroby w większej populacji pacjentów, określenie związku pomiędzy jego bezpieczeństwem, a skutecznością podczas krótkotrwałego i długotrwałego stosowania. Ma ona również potwierdzić wyniki dotyczące skuteczności leku uzyskane w fazie II. Pozwala także na poznanie rzadszych działań ubocznych, zdobycie danych koniecznych do dopuszczenia leku do powszechnego stosowania. Ta część badań może trwać od roku do kilku lat. Zasady prowadzenia III fazy badań (zastosowanie metody podwójnie ślepej próby, losowy dobór pacjentów do poszczególnych grup) są takie same, jak w fazie II, wymaga również częstych wizyt w klinice prowadzącej badania.
W tej fazie badania porównywanie działania badanego leku (LB) z placebo (P), albo z lekiem standardowym (LS), wykonywane może być na trzy sposoby:
Badania tej fazy zbierają dane, które są podstawą do rejestracji produktu leczniczego (faza III a) oraz służą celom marketingowym (faza III b). Po pozytywnym zakończeniu III fazy badań lek może zostać zarejestrowany i wprowadzony do obrotu. Na podstawie wyników fazy: III a oraz III b następuje przygotowanie wniosku o rejestrację nowego produktu leczniczego.
Wszystkie dane uzyskane w czasie badań podstawowych, przedklinicznych oraz klinicznych fazy od I do III są obowiązkowym elementem dokumentacji, wymaganej przez instytucje zajmujące się rejestracją leków. Dokumentacja rejestracyjna może liczyć nawet kilkanaście tysięcy stron. Przed rozpoczęciem procesu badawczo-rozwojowego producent produktu leczniczego konsultuje z wiodącymi, instytucjami rejestracji leków zakres niezbędnych danych, w tym kryteria włączenia i wyłączenia oraz wskazania, aby zminimalizować ryzyko odrzucenia dokumentacji rejestracyjnej z powodu pominięcia istotnych danych.
III faza badań klinicznych trwa od roku do kilku lat, i jest prowadzona z udziałem aż kilku tysięcy chorych (od 1 000 do 3 000 i więcej). Pacjenci kwalifikowani do badań III fazy to chorzy ze wskazaniem do farmakoterapii, kwalifikowani są do badania na podstawie ścisłych kryteriów mających na celu przebadanie leku w populacji zbliżonej do populacji chorych występującej w praktyce medycznej. Prowadzona jest ona zwykle przez wiele ośrodków klinicznych, często w wielu krajach. Liczba pacjentów włączanych do badania uwarunkowana jest wymogami rejestracyjnymi (FDA lub EMEA) oraz statystycznymi (szacowana wielkość próby).
Faza III ma na celu ostateczne potwierdzenie skuteczności badanej substancji w leczeniu danej choroby w większej populacji pacjentów, określenie związku pomiędzy jego bezpieczeństwem, a skutecznością podczas krótkotrwałego i długotrwałego stosowania. Ma ona również potwierdzić wyniki dotyczące skuteczności leku uzyskane w fazie II. Pozwala także na poznanie rzadszych działań ubocznych, zdobycie danych koniecznych do dopuszczenia leku do powszechnego stosowania. Ta część badań może trwać od roku do kilku lat. Zasady prowadzenia III fazy badań (zastosowanie metody podwójnie ślepej próby, losowy dobór pacjentów do poszczególnych grup) są takie same, jak w fazie II, wymaga również częstych wizyt w klinice prowadzącej badania.
W tej fazie badania porównywanie działania badanego leku (LB) z placebo (P), albo z lekiem standardowym (LS), wykonywane może być na trzy sposoby:
- poprzez porównanie dwóch grup chorych, z których jedna otrzymuje LB, a druga P lub LS,
- parami, kiedy jeden chory otrzymuje LB, a drugi w tym samym czasie P lub LS,
- krzyżowo- wtedy, gdy jeden chory najpierw otrzymuje LB, potem P lub LS i znowu LB, a drugi chory leczony jest w odwrotnej kolejności - najpierw P lub LS, potem LB i znowu P lub LS.
Badania tej fazy zbierają dane, które są podstawą do rejestracji produktu leczniczego (faza III a) oraz służą celom marketingowym (faza III b). Po pozytywnym zakończeniu III fazy badań lek może zostać zarejestrowany i wprowadzony do obrotu. Na podstawie wyników fazy: III a oraz III b następuje przygotowanie wniosku o rejestrację nowego produktu leczniczego.
Wszystkie dane uzyskane w czasie badań podstawowych, przedklinicznych oraz klinicznych fazy od I do III są obowiązkowym elementem dokumentacji, wymaganej przez instytucje zajmujące się rejestracją leków. Dokumentacja rejestracyjna może liczyć nawet kilkanaście tysięcy stron. Przed rozpoczęciem procesu badawczo-rozwojowego producent produktu leczniczego konsultuje z wiodącymi, instytucjami rejestracji leków zakres niezbędnych danych, w tym kryteria włączenia i wyłączenia oraz wskazania, aby zminimalizować ryzyko odrzucenia dokumentacji rejestracyjnej z powodu pominięcia istotnych danych.

- EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Re: EVR- wycena 29 zł +67ze kardioznacznik
- Kurs Euro
- Kurs dolar
- Kurs frank
- Kurs funt
- Wiron
- Przelicznik walut
- Kantor internetowy
- Kalkulator wynagrodzeń
- Umowa zlecenie
- Kredyt na mieszkanie
- Kredyt na samochód
- Kalkulator kredytowy
- Revolut
- Winiety
- Jak grać na giełdzie?
- Jak wziąć kredyt hipoteczny?
- Rejestracja samochodu
- Jak rozwiązać umowę z Orange
- Koszty uzyskania przychodów
- Sesje elixir
- PB weekend
- RRSO co to jest?
- Blogbank.pl
- Promocje bankowe - zgarnijpremie.pl
- Stopa procentowa